基本信息
联系方式
机构简介
机构概况
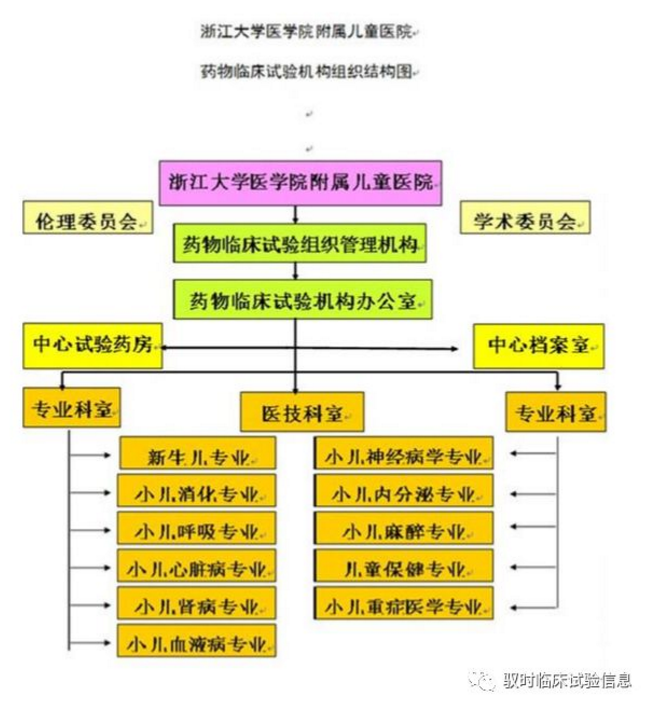
浙江大学医学院附属儿童医院药物临床试验机构组织管理机构接受浙江大学医学院附属儿童医院院长直接领导,接受国家卫计委和国家食品药品监督管理总局的双重监督。
本院药物临床试验机构组织管理机构负责人由分管院长直接担当,下设临床试验机构办公室,其主要负责对各专业药物临床试验进行统一管理、监督和指导,以保证临床试验严格按GCP规范执行。药物临床试验机构组织管理机构接受专家委员会的指导。临床试验机构办公室统一配备了带锁文件柜、联网计算机、直拨电话、传真机、打印机、复印机等。临床试验机构环境整洁,有空调和消防灭火设施,能够较好满足临床试验的需要。
临床试验机构办公室下设中心档案室、中心试验药房、专业科室和医技科室。
中心档案室、中心试验药房分别管理试验文档和试验药物,并且指派专业且经受过培训人员进行管理。中心档案室配备了带锁文件柜、联网计算机、刻录机等,环境整洁,通风性较好、安装有空调和灭火器,能够满足临床试验的需要。中心试验药房配备了带锁药品柜、带锁冰箱、温湿度计等,环境整洁,安装有空调调控温度,通风性较好、消防设施齐全,能够满足临床试验的需要。
医技科室也纳入整个药物临床试验管理范畴,医技科室主要包括:中心实验室、特检科、放射科、检验科、临床药学实验室等,目前这些科室均已建立了完善的管理制度和标准操作规程,并且指派了医技科室专业负责人直接参与临床试验管理。
机构于2014年10月21日通过药物临床试验专业复核认证,认证专业为小儿消化、小儿呼吸、小儿心脏病、小儿肾病、小儿血液病、小儿神经病学、小儿内分泌、儿童保健、小儿重症医学、小儿麻醉。
专业科室均设立了研究者办公室和受试者接待室。各申请专业的病房与ICU室处均有绿色通道相通,能够尽最大力量充分保护受试者的安全。
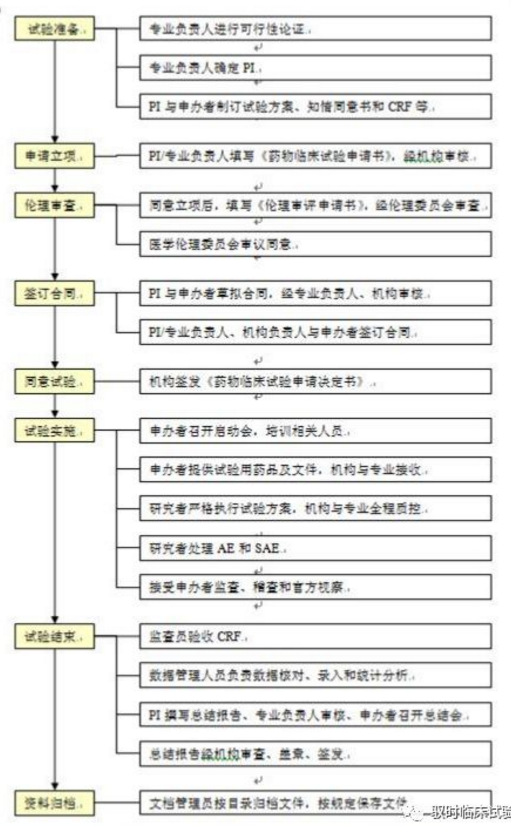
临床试验工作流程
临床试验资料递交清单
药物临床试验送审材料清单
□ 药物临床试验申请书
□ 申办者委托书(委托主要研究者、CRO、CRA/CRC)
□ 药物临床试验批件/注册批件
□ 申办者/CRO资质(营业执照、生产许可证、组织机构代码证和法人身份证复印件等)
□ 药检报告
□ 临床试验方案
□ 研究者手册
□ CRF及原始病历空白表单
□ 知情同意书(ICF)
□ 研究小组及分工
□ 中心伦理批件
□ 临床研究协议草案
□ 其他向受试者提供的书面材料
□“药物临床试验登记与信息公示平台”登记的证明或描述文件
医疗器械临床试验送审材料清单
□ 医疗器械临床试验申请书
□ 申办者委托书 (委托主要研究者、CRO、CRA/CRC)
□ 申办者/CRO资质(营业执照、生产许可证、组织机构代码证和法人身份证复印件等)
□ 注册产品标准或相应的国家、行业标准
□ 产品型式试验报告
□ 产品自测报告(包括对照品)
□ 动物试验报告(需要时)
□ 试验研究方案
□ 病例报告表(CRF)
□ 知情同意书(ICF)
□ 医疗器械临床试验须知
□ 研究产品的临床背景资料
□ 向医疗机构提供的担保
□ 研究小组及分工
□ 临床研究协议
□ 其他(如其它中心对本研究的伦理情况,招募广告,日记卡或其他向受试者提供的书面材料)
体外诊断试剂临床试验送审材料清单
□ 体外诊断试剂临床试验申请书
□ 申办者委托书(委托主要研究者、CRO、CRA/CRC)
□ 申办者/CRO资质(营业执照、生产许可证、组织机构代码证和法人身份证复印件等)
□ 注册产品标准或相应的国家、行业标准
□ 试剂质量检查报告(包括对照试剂)
□ 试验研究方案
□ 研究者手册
□ 病例报告表(CRF)(需要时)
□ 知情同意书(ICF)(需要时)
□ 主要研究者简历、研究小组及分工
□ 研究协议
□ 其他(如其它中心对本研究的伦理情况等)
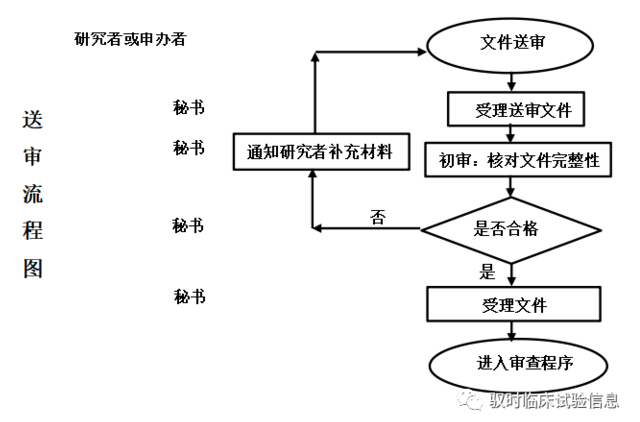
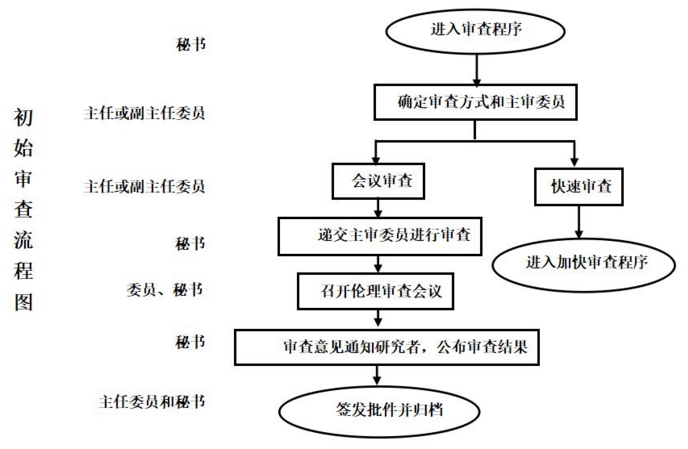
伦理审查及受理流程
项目工作流程
临床试验工作流程
立项资料递交
Submitted Files for Review
药物临床试验初始审查申请文件清单:
1 | 送审项目资料清单(注明所有递交文件的版本号和日期) |
2 | 同意立项通知函 |
3 | 初始审查申请书(申请者签名并注明日期) |
4 | 药物临床试验批件/临床试验通知书 /默示许可相关资料(或者药品注册批件) |
5 | 申办者/CRO资质:营业执照\生产许可证\法人身份证复印件等 |
6 | 申办者委托书(委托主要研究者、CRO、CRA) |
7 | 临床试验方案(注明版本号/日期,申办者、研究者已签字) |
8 | 病历报告表样表(注明版本号/日期) |
9 | 研究者手册(注明版本号/日期) |
10 | 受试者知情同意书(注明版本号/日期) |
11 | 试验药物质量检查报告(包括对照药物) |
12 | 研究小组名单及分工、研究人员简历(含GCP证书) |
13 | 研究协议草案 |
14 | 现有的安全性资料(如有) |
15 | 受试者招募广告(如有,需注明版本号/日期) |
16 | 受试者保险的相关文件(如有) |
17 | 其他提供给受试者的任何书面资料(如有) |
18 | 其他(如:组长单位伦理审查批件、“药物临床试验登记与信息公示平台”登记证明或描述文件、“人类遗传资源相关情况的声明”等) |
医疗器械临床试验初始审查申请文件清单:
1 | 送审文件清单(注明所有递交文件的版本号和日期) |
2 | 同意立项通知函 |
3 | 初始审查申请书(申请者签名并注明日期) |
4 | 申办者委托书(委托主要研究者、CRO、CRA/CRC) |
5 | 申办者/CRO资质(营业执照和生产许可证等) |
6 | 注册产品标准或相应的国家、行业标准 |
7 | 产品型式试验报告 |
8 | 产品自测报告(包括对照品) |
9 | 动物试验报告(需要时) |
10 | 试验研究方案(注明版本号/日期) |
11 | 病例报告表(CRF)(注明版本号/日期) |
12 | 知情同意书(ICF)(注明版本号/日期) |
13 | 临床试验机构的设施和条件能够满足试验的综述 |
14 | 试验用医疗器械的研制符合适用的医疗器械质量管理体系相关要求的声明 |
15 | 研究者手册/医疗器械临床试验须知(如有) |
16 | 研究产品的临床背景资料(如有) |
17 | 受试者保险的相关文件或向医疗机构提供的担保 |
18 | 研究小组名单及分工、研究人员简历(含GCP证书) |
19 | 研究协议草案 |
20 | 其他(如:组长单位的伦理审查情况,招募广告,日记卡,人类遗传资源相关声明或其他向受试者提供的书面材料) |
体外诊断试剂初始审查申请文件清单::
1 | 送审文件清单(注明所有递交文件的版本号和日期) |
2 | 初始审查申请书(申请者签名并注明日期) |
3 | 同意立项通知函 |
4 | 申办者委托书(委托主要研究者、CRO、CRA/CRC) |
5 | 申办者/CRO资质(营业执照和生产许可证等) |
6 | 注册产品标准或相应的国家、行业标准 |
7 | 试剂质量检查报告(包括对照试剂) |
8 | 试验研究方案(注明版本号/日期) |
9 | 研究者手册(注明版本号/日期) |
10 | 病例报告表(CRF)(需要时,并注明版本号/日期) |
11 | 知情同意书(ICF)(需要时,并注明版本号/日期) |
12 | 研究小组名单及分工、研究人员简历(含GCP证书) |
13 | 研究协议草案 |
14 | 其他(如组长单位的伦理审查情况,人类遗传资源相关声明等) |
修正后复审伦理审查申请文件清单:
1 | 送审文件清单目录(注明所有递交文件的版本号和日期) |
2 | 伦理审评意见回复函 |
3 | 各文件修正前后对照表(注明修正位置、修改前内容及修改后的内容)或痕迹版文件(注明版本号和日期,所作修改处必须划线或阴影标示) |
4 | 相关文件清洁版(注明版本号和日期) |
修正案审查申请文件清单:
1 | 送审文件清单目录(注明所有递交文件的版本号和日期) |
2 | 修正案申请表 |
3 | 各文件修正前后对照表(注明修正位置、修改前内容及修改后的内容)或痕迹版文件(注明版本号和日期,所作修改处必须划线或阴影标示) |
4 | 相关文件清洁版(注明版本号和日期) |
年度/定期跟踪审查申请文件清单:
1 | 年度/定期跟踪审查申请表 |
2 | 研究过程中发生情况的详细说明(如需要) |
严重不良件事件应递交的文件清单:
1 | 严重不良事件报告表 |
2 | 外院SAE还应递交安全性事件或报告研究者评估表. |
药物临床试验SUSAR或安全性报告应递交的文件清单:
1 | SUSAR或安全性报告 |
2 | 安全性事件或报告研究者评估表. |
违背方案报告应递交的文件清单:
1 | 违背方案报告 |
2 | 违背方案的详细情况列表(如有) |
暂停/终止研究申请文件清单:
1 | 暂停/终止研究申请表 |
2 | 小结报告(如有) |
结题报告审查申请文件清单:
1 | 结题申请表 |
2 | 分中心小结表/研究总结报告 |
3 | 发表文章(如有) |
4 | 临床试验人类遗传资源管理调查表(如需) |
5 | “人类遗传资源数据备份平台”数据备份完成证明文件或相关说明(如需) |
注意事项:
1. 所有材料请提供纸质版2份和电子版1份,递交纸质文件前,请先在我院“临床试验管理平台”上递交相关文件,并将电子版文件发送至邮箱:zuchiec@163.com。
2. 申请表必须递交原件,并有签名和日期。
初次递交文件时,需使用 A4双孔活页夹(参考型号:齐心A 205),可根据文件多寡选择文件夹厚度,不同文件之间要用隔页纸,文件夹内要附目录,外(正侧面)要有项目名称标识;后续递交文件需打双孔后递交。
申请编号(机构编写): 递交日期: 年 月 日
药物临床试验申请书
浙江大学医学院附属儿童医院药物临床试验机构办公室:
现有一项药物临床试验拟在我院_________(专业)开展,题目为:
__________________________________________
试验药通用名:
试验药类别:中药、天然药物 【 】 类
化学药物 【 】 类 【 】项
治疗用生物制品 【 】 类
预防用生物制品 【 】 类
上市药物再评价 【 】
试验分期:
□ I期(仅适用于1、2类药) □ II期(仅适用于1、2类药)
□ III期(仅适用于1、2类药) □ IV期(仅适用于1、2类药)
□ 临床试验 □ 药代动力学 □ 生物等效性
申办公司名称 | CRO名称 |
|
|
申办公司地址 | CRO地址 |
|
|
申办者代表及联系电话 | CRO代表及联系电话 |
|
|
附件材料(请逐项核实)
□ 申办者委托书(委托主要研究者、CRO、CRA/CRC)
□ 药物临床试验批件/临床试验通知书/默示许可相关资料 (或者药品注册批件)
□ 申办者/CRO资质 (营业执照、生产许可证和法人身份证复印件等)
□ 试验药物质量检查报告(包括对照药物)
□ 试验研究方案
□ 研究者手册
□ 病例报告表(CRF)及其他相关文件(样稿)
□ 知情同意书(ICF)
□ 研究人员简历和研究小组名单及分工
□ 组长单位伦理批件(本机构为非组长单位的项目适用)
□ 临床研究协议草案
□“药物临床试验登记与信息公示平台”登记的证明或描述文件(如需)
□“人类遗传资源”相关情况的声明(如需)
□ 其他(如招募广告,日记卡或其他向受试者提供的书面材料等)
研究承诺:
我已阅读试验研究方案及相关材料,若该项目获得批准,我将负责该临床试验全过程中的质量保证,承诺该临床试验数据真实可靠,操作规范,符合国家药品监督管理部门《药物临床试验质量管理规范》(GCP)要求。如有失实,愿意承担相关责任。
主要研究者签名: 日期: 年 月 日
![]()
专业负责人意见:
专业负责人签名: 日期: 年 月 日
![]()
说明:
申办者委托书、临床试验批件/默示许可相关资料或药品注册批件、资质证书、质量检查报告、其他中心伦理批件等需加盖单位红章,有多页者需加盖骑缝章或每页盖章。
CRO指提供临床试验服务的合同研究公司,CRA是指监查员, CRC是指研究助理或协调员。
申办者委托的CRA/CRC需附身份证复印件、毕业证书、GCP培训证书复印件。
若为境外企业,可以用办事人身份证复印件替代法人身份证复印件递交。
用黑色或蓝黑色钢笔填写此表,字迹要清楚、工整,不得涂改。
如有【 】,请申办者对照《药品注册管理办法》有关药品注册分类填写,在相应栏目上标注相应的数字。
如有□请在相应栏目上画√。
请查看内容是否有遗漏。
研究小组及分工
临床研究题目___________________________________________________________________
申办单位 ___________________________________________________________________
研究协作中心*1.组长单位: 2.
3. 4.
| |||||
研究人员名单(请填写所有参加研究的人员信息) | |||||
研究者 | 姓名 | 职称 | 联系电话 | 分工 | 是否经过GCP培训 |
专业负责人 |
|
|
|
|
|
主要研究者 (PI) |
|
|
|
|
|
协作研究者 |
|
|
|
|
|
协作研究者 |
|
|
|
|
|
研究护士 |
|
|
|
|
|
研究护士 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
分工: 1. 本试验的秘书(负责与机构、伦理委员会等的联系) 2. 病人积累 3. 知情同意 4. 体检及询问病史 5. 入选/排除标准的判断 6. 指导用药 7. 科室药物管理(至少2人) |
8. SAE汇报 9. 填写CRF表 10. CRF表更正/差异的修改 11. 实验室检查联系 12. 试验文档管理(至少2人) 13. 质量控制(至少1人) 14. 病人随机化 15. 试验评估 16. 其他 | ||||
主要研究者签字: 日期
医疗器械临床试验申请书
浙江大学医学院附属儿童医院临床试验机构办公室:
现有一项医疗器械临床试验拟在我院_________(专业)开展,题目为:___________________________________________
医疗器械类别: □ 二 类 □ 三类
试验类别: □ 临床试用 □ 临床验证
申办公司名称 | CRO名称 |
|
|
申办公司地址 | CRO地址 |
|
|
申办者代表及联系电话 | CRO代表及联系电话 |
|
|
附件材料(请逐项核实)
□申办者委托书 (委托研究中心、主要研究者、CRO、CRA/CRC)
□申办者/CRO资质[营业执照(三证合一)、生产许可证(如有)、法人身份证复印件等]
□注册产品标准或相应的国家、行业标准
□产品型式试验报告
□产品自测报告(包括对照品)
□产品注册检验报告
□动物试验报告(需要时)
□试验研究方案
□病例报告表(CRF)
□知情同意书(ICF)
□研究者手册(IB)
□向医疗机构提供的担保
□主要研究者简历、研究小组及分工
□临床研究协议初稿
□人类遗传资源相关声明
其他(如组长单位对本研究的伦理情况,招募广告,日记卡,临床试验保险凭证或其他向受试者提供的书面材料)
研究承诺:
我已阅读试验研究方案及相关材料,若该项目获得批准,我将负责该临床试验全过程中的质量保证,承诺该临床试验数据真实可靠,操作规范,符合相应规范要求。如有失实,愿意承担相关责任。
主要研究者签名: 日期: 年 月 日
![]()
专业负责人意见:
专业负责人签名: 日期: 年 月 日
说明:
申办者委托书、NMPA临床试验批件或医疗器械注册批件、资质证书、质量检查报告、组长单位伦理批件等需加盖单位红章,有多页者需加盖骑缝章或每页盖章。
CRO指提供临床试验服务的合同研究公司,CRA是指监查员, CRC是指研究助理或协调员。
申办者委托的CRA/CRC需附身份证复印件、毕业证书、GCP培训证书复印件。
若为境外企业,可以用办事人身份证复印件替代法人身份证复印件递交。
用黑色或蓝黑色钢笔填写此表,字迹要清楚、工整,不得涂改。
医疗器械类别,请申办者对照《医疗器械注册管理办法》和《医疗器械监督管理条例》有关医疗器械注册分类填写。
如有□请在相应栏目上画√。
本表一式两页(不包括填表说明页),请查询是否有遗漏。
申请编号(机构编写): 递交日期: 年 月 日
体外诊断试剂临床试验申请书
浙江大学医学院附属儿童医院药物临床试验机构办公室:
现有一项体外诊断试剂临床研究拟在我院_________(专业)开展,题目为:___________________________________________
试剂名称:
试剂分类: □ 境内 【 】 类体外诊断试剂
□ 境外体外诊断试剂
申办公司名称 | CRO名称 |
|
|
申办公司地址 | CRO地址 |
|
|
申办者代表姓名及联系电话 | CRO代表姓名和联系电话 |
|
|
附件材料(请逐项核实)
□ 申办者委托书 (委托主要研究者、CRO、CRA/CRC)
□ 申办者/CRO资质(营业执照、生产许可证、法人身份证复印件等)
□ 注册产品标准或相应的国家、行业标准
□ 试剂质量检查报告(包括对照试剂)
□ 试验研究方案
□ 研究者手册
□ 病例报告表(CRF)(需要时)
□ 知情同意书(ICF)(需要时)
□主要研究者简历、研究小组及分工
□ 临床研究协议初稿
□人类遗传资源相关声明
□ 其他(如组长单位对本研究的伦理情况等)
研究承诺:
我已阅读试验研究方案及相关材料,若该项目获得批准,我将负责该临床试验全过程中的质量保证,承诺该临床试验数据真实可靠,操作规范,符合相关规范要求。如有失实,愿意承担相关责任。
主要研究者签名: 日期: 年 月 日
![]()
专业负责人意见:
专业负责人签名: 日期: 年 月 日
![]()
填表说明:
申办者委托书、申办者资质证书、注册产品标准或相应的国家和行业标准、质量检查报告、组长单位的伦理批件等需加盖单位红章,有多页者需加盖骑缝章或每页盖章。
CRO指提供临床试验服务的合同研究公司,CRA是指监查员, CRC是指研究助理或协调员。
申办者委托的CRA/CRC需附身份证复印件、毕业证书、GCP培训证书复印件。
若为境外企业,可以用办事人身份证复印件替代法人身份证复印件递交。
用黑色或蓝黑色钢笔填写此表,字迹要清楚、工整,不得涂改。
如有【 】,请申办者对照《体外诊断试剂注册管理办法》有关体外诊断试剂注册分类填写,在相应栏目上画√,标注相应的数字。
如有□请在相应栏目上画√。
CRO指提供临床试验服务的合同研究公司。
本表一式两页(不包括填表说明页),请查询是否有遗漏。
体外诊断试剂研究小组及分工
临床研究题目___________________________________________________________________
申办单位 ___________________________________________________________________
研究协作中心*1.组长单位: 2.
3. 4.
| |||||
研究人员名单(请填写所有参加研究的人员信息) | |||||
研究者 | 姓名 | 职称 | 联系电话 | 分工 | 是否经过GCP培训 |
专业负责人 |
|
|
|
|
|
主要研究者 (PI) |
|
|
|
|
|
协作研究者 |
|
|
|
|
|
协作研究者 |
|
|
|
|
|
研究护士 |
|
|
|
|
|
研究护士 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
分工: 1. 本试验的秘书(负责与机构、伦理委员会等的联系) 2. 病人积累 3. 知情同意 4. 体检及询问病史 5. 入选/排除标准的判断 6. 试剂使用指导 7. 科室试剂管理(至少2人) |
8. SAE汇报 9. 填写CRF表 10. CRF表更正/差异的修改 11. 实验室检查联系 12. 试验文档管理(至少2人) 13. 质量控制(至少1人) 14. 病人随机化 15. 试验评估 16. 其他 | ||||
主要研究者签字: 日期
其他情况
以上信息如有错误,
请卿长按二维码进入公众号留言正确信息,
蔽号晓筑守12小时内修改更新,
感谢卿的支持!

